
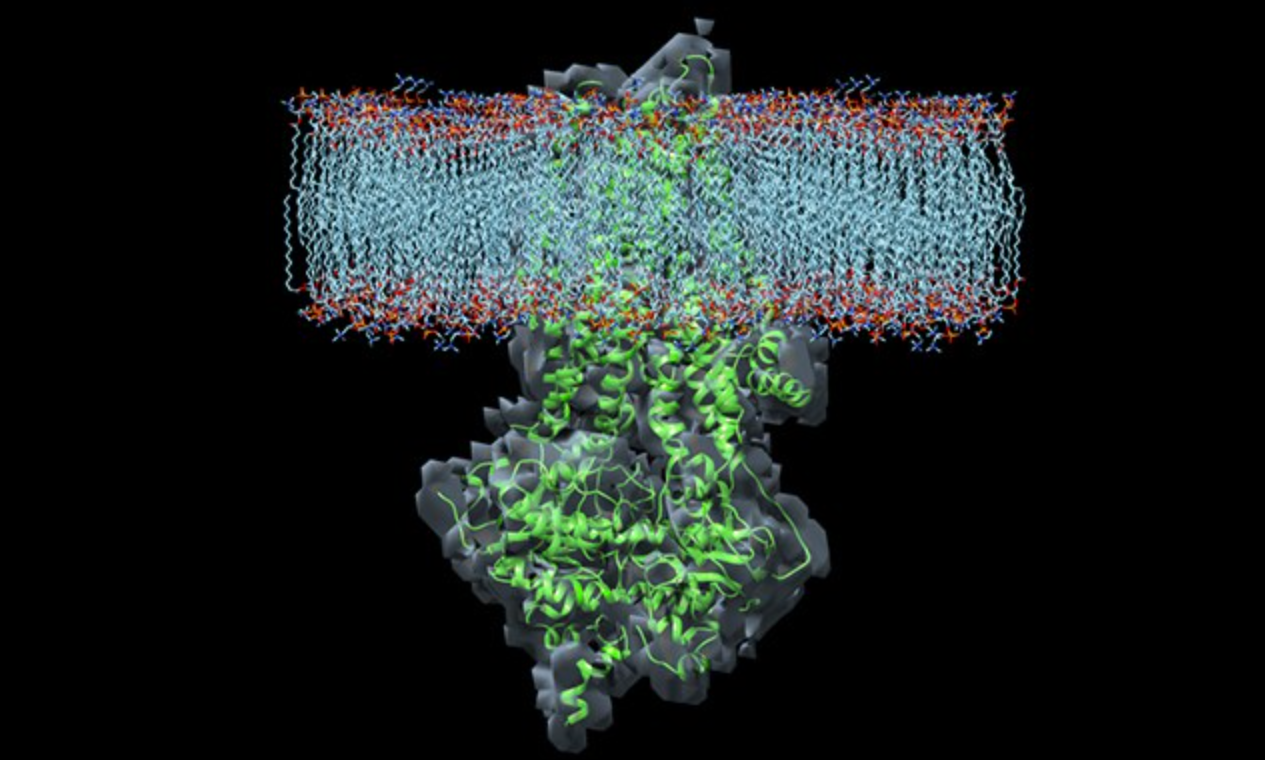
Is there a selective advantage to being heterozygous for a cystic fibrosis gene mutation? If so, what does this mutation protect against? These – and other questions – were discussed in Episode 21 of the podcast. But, before getting into potential answers, some background is in order.
Cystic fibrosis is an autosomal recessive genetic disorder where lack of production or dysfunction of an epithelial chloride channel called the cystic fibrosis transmembrane conductance regulator (CFTR) leads to recurrent sinopulmonary infections, bronchiectasis and structural lung damage, pancreatic insufficiency, and other symptoms. It is most common in white people of European descent (1 in 3,000) with 1 in 30 being carriers, and is much less frequent in other populations.
Does sickle cell disease provide a clue?
Many are familiar with the fact that heterozygous carriers of the mutation for sickle cell disease (i.e., those with sickle cell trait) are protected against malaria. This helps explains the prevalence of a gene that otherwise would be selected against. Could something similar explain the prevalence of cystic fibrosis?
Maybe.
As will be discussed below, there is data suggesting that the prevalence of cystic fibrosis gene mutations may be explained by protection against infection by Mycobacterium tuberculosis (MTB). While infection by nontuberculous mycobacteria is relatively common in patients with cystic fibrosis, infection with MTB is exceedingly rare. It is so rare that encountering a patient with cystic fibrosis with MTB in clinical practice is case-reportable. That’s literally what the published case reports write: Look! This patient with cystic fibrosis somehow got tuberculosis.
Where’s the evidence?
When historians examine whether carriers of a genetic disease have a potential selective advantage they use three types of evidence.
- Historical and/or Geographical
- Clinical or Population-based
- Molecular
Historical and/or Geographical Evidence
MTB is a really, really old affliction. It has been found in bison remains from 17,000 years ago and from pre-historic human skeletons from 4000 BC. In the 1600s an epidemic of MTB epidemic known as the White Plague spread across most of Europe. Over the next 300 years the White Plague caused up to a quarter of all deaths. It was called the White Plague because of the wan complexion of many of the patients dying of the disease
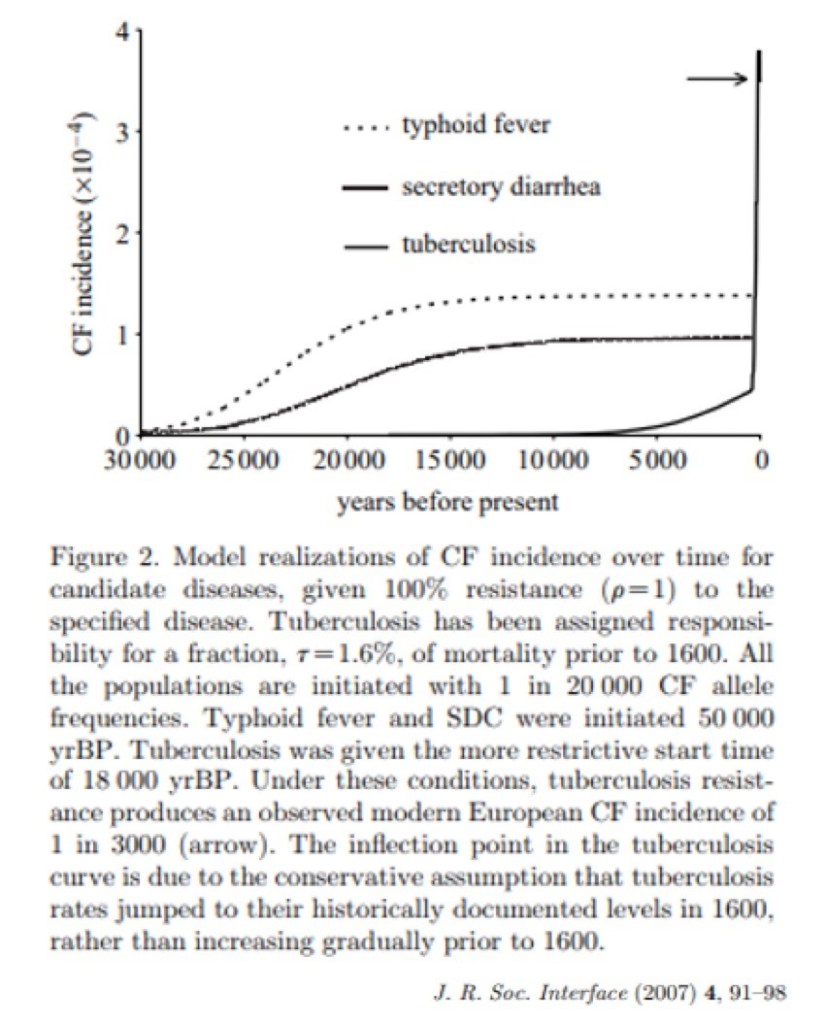
In 2007 a group examined the hypothesis that being heterozygous for a cystic fibrosis mutation is protective against infectious diseases. In particular, they looked to see if any infections might account for modern prevalence of the carrier rate of cystic fibrosis mutations (recall that’s about 1:30 in white Europeans).
The researchers studied the current prevalence of MTB, typhoid, and cholera and looked backward in time. In the end, they found that only a significant spike in MTB prevalence around the year 1600 would account for current rates of cystic fibrosis carriers in current populations. This matches the timing of the White Plague pandemic discussed above. These findings suggest that the current prevalence of the cystic fibrosis trait is explained by protection if offers against MTB infection.
The gradual growth in cholera and typhoid rates over that time would not account for the current prevalence of cystic fibrosis gene mutations.
Clinical or Population-based evidence
A group of researchers from Brazil examined the rates of cystic fibrosis heterozygosity and compared this with MTB incidence in the same areas. They found that MTB incidence was inversely proportional to the rates of cystic fibrosis trait. This was true even after controlling for factors such as socioeconomic status.
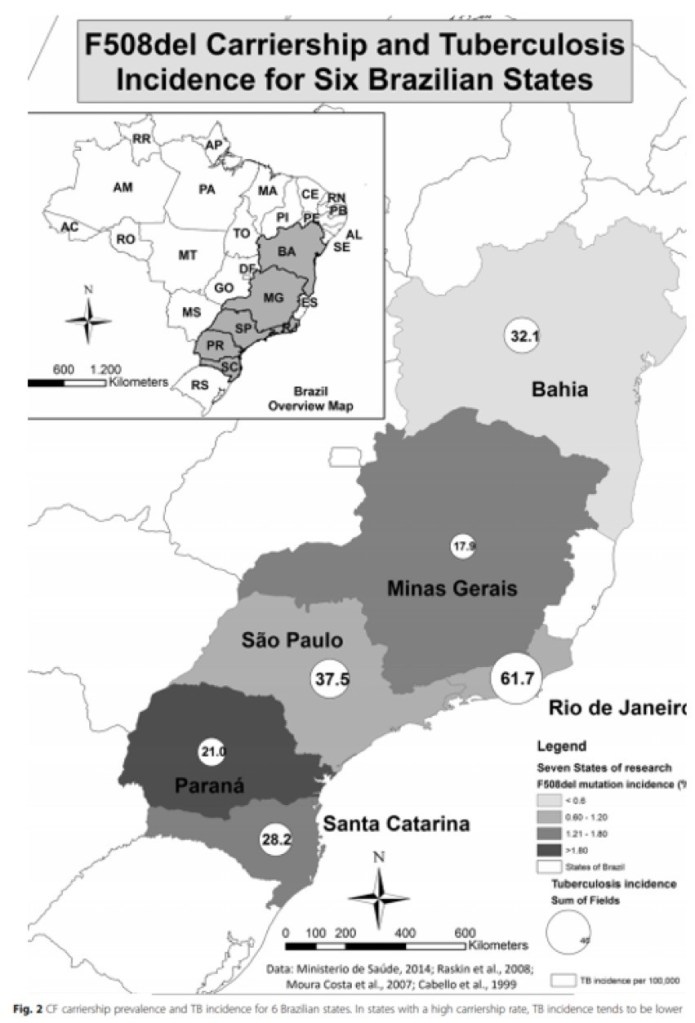
Interestingly, there is also literature from the 1970s suggesting that parents of patients with cystic fibrosis (who are heterozygous carriers) have reduced rates of TB infection as well.
Molecular evidence
While the first two forms of evidence are thought-provoking, a mechanistic explanation would lend credence to the hypothesis that a rise in MTB created selective pressure for cystic fibrosis mutations. Does such an explanation exist? Yes!
Recall that mycobacterium tuberculosis has a complex cell wall and that some of the components are glycolipids, including phosphatidylinositol mannoside and mannose-capped lipoarabinomannan.

These glycolipids help MTB avoid destruction by the immune system, specifically mediators such as alveolar macrophages. When the bacterium is taken up by macrophages, it gets sequestered in phagolysosomes and is ultimately destroyed. The glycolipids help prevent the phagolysosomes from fusing, which is a crucial step in destroying the bacteria. They are an important component of the virulence of the organism.
But the glycolipids have to be in a specific state to effectively block phagolysosome function. More specifically, they must be sulfated (i.e. have a sulfate group added). Sulfation of glycolipids helps prevent MTB from destruction, and is a crucial component of TB successfully infecting people.
But MTB has a problem. It lacks arylsulfatase B, the enzyme needed to isolate sulfate groups. Humans – on the other hand – do have arylsulfatase B! Under normal circumstances our own arylsulfatase B will provide the sulfate groups for MTB to use. It then can block phagolysosome function and cause infection. MTB uses our own machinery against us.
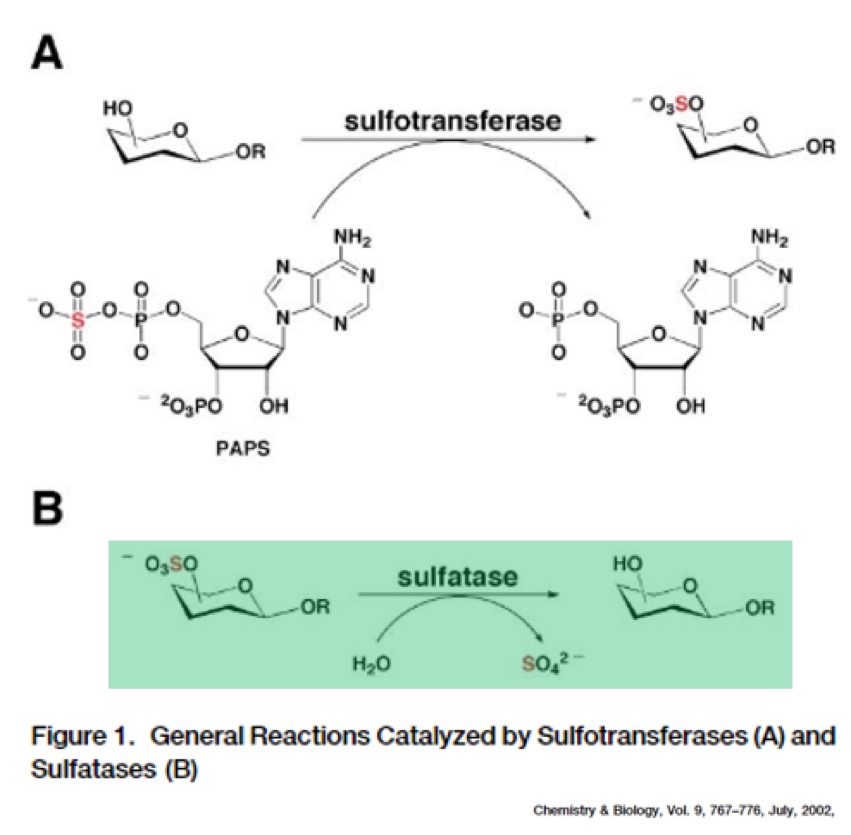
Here’s the key: patients with cystic fibrosis have significantly decreased levels of activity of arylsulfatase B! This probably results from higher intracellular chloride levels when CFTR is dysfunctional, which disrupts trafficking of the arylsulfatase B enzyme protein inside cells. The result? People with cystic fibrosis may be protected from MTB infection due to decreased levels of the arylsulfatase B enzyme and reduced access of the organism to sulfate groups. Unfortunately, the relative function of arylsulfatase B in CF carriers has not been studied.
Interestingly, there is also data suggesting that this mechanism may contribute to susceptibility to pseudomonas colonization and infection seen in patients with cystic fibrosis. Pseudomonas possesses its own sulfatase enzyme. It therefore can metabolize sulfated esters, liberating sugars which can be used for fuel.
What about cholera?
Many assume that cystic fibrosis mutations protect against cholera. Theoretically it would make sense that cystic fibrosis mutations could protect against cholera since the secretory diarrhea is mediated via CFTR and chloride secretion. And while it is possible that protection from cholera, or typhoid for that matter, could account for some component of cystic fibrosis prevalence, there are important arguments against cholera being the dominant selective driver.
First is the population level, modeling data discussed above. It shows that the current rates of cystic fibrosis mutations in the population are not accounted for by historical rates of cholera infection.
Second, there actually is evidence against the cholera hypothesis. When exposed to a cholera toxin analogue, intestinal cells of cystic fibrosis heterozygotes secrete just as much chloride as wild type controls, arguing against a selective advantage for carriers. But that being said, intestinal cells from people who have clinical CF have essentially zero intestinal chloride secretion to begin with and this did not increase with exposure to the analogue of the cholera toxin.
Take Home Points
- Patients with cystic fibrosis appear to be protected from Mycobacterium tuberculosis
- There is some retrospective, population-based data suggesting that cystic fibrosis carriers may be protected as well and that an extended Mycobacterium tuberculosis epidemic in Europe from the 1600s-1900s might account for the prevalence of cystic fibrosis mutations in current populations
- The mechanism of protection is theorized to be deficiency of the arylsulfatase B enzyme, which interferes with one of the main virulence factors of Mycobacterium tuberculosis.
CME/MOC
Click here to obtain AMA PRA Category 1 Credits™ (1.00 hours), Non-Physician Attendance (1.00 hours), or ABIM MOC Part 2 (1.00 hours).
Listen to the episode
Credits & Citation
◾️Episode written by Avi Cooper
◾️Show notes written by Tony Breu and Avi Cooper
◾️Audio edited by Clair Morgan of nodderly.com
Cooper AZ, Abrams HR, Breu AC. Why is cystic fibrosis one of the most common genetic diseases? The Curious Clinicians Podcast. March 31, 2021.
Image credit: https://www.news4jax.com/news/local/2020/04/04/thermometers-flying-off-store-shelves-during-pandemic/
Related tweetorial: https://x.com/avrahamcoopermd/status/1224814287379562496

One thought on “Episode 22: Why is cystic fibrosis one of the most common genetic diseases?”