

Why can heparin cause hyperkalemia?
Most of us have seen hospitalized patients with mild but persistent hyperkalemia with no obvious cause. You notice a potassium of 5.3-5.5 mmol/L every day, even though the patient had a normal potassium level on admission. These patients are not on spironolactone, and their renal function is normal. There just isn’t a good explanation for their elevated serum potassium levels.
Or is there?
Turns out that in many of these cases the culprit is the heparin or enoxaparin venous thromboembolism prophylaxis we administer.
Heparin was discovered in 1916 by a medical student at Johns Hopkins named Jay McLean. He was studying dog livers and isolated a fat-soluble glycosaminoglycan anticoagulant. Because it was originally found in the liver it was named heparin, acknowledging the Greek word for liver (hepar). But heparin is made naturally all over the body, not just by the liver. Mast cells are the main, and some theorize only, producers of heparin in the body. It gets released from their secretory granules at sites of tissue injury.
The association between heparin and hyperkalemia was first observed in 1964 when a patient with recurrent myocardial infarctions received a treatment course lasting a few weeks. He developed hyperkalemia, with a peak serum potassium level of 6.4 mmol/L. The authors did not have a good explanation for it other than the heparin.
Subsequent case reports and series show this association, both with unfractionated heparin and low molecular weight heparin. The exact rate is not known, but a review from 1995 in the American Journal of Medicine estimated the incidence of mild hyperkalemia with heparin exposure to be approximately 7%. Severe hyperkalemia requires other factors to be present (e.g., other medications known to raise potassium or renal dysfunction). A similar picture is seen with low molecular weight heparin exposure. A small, prospective study from 2011 in the Journal of Pharmacology and Pharmacotherapeutics found that about 9% of patients treated with low molecular weight heparin developed mild hyperkalemia, with the main risk factor being impaired kidney function.
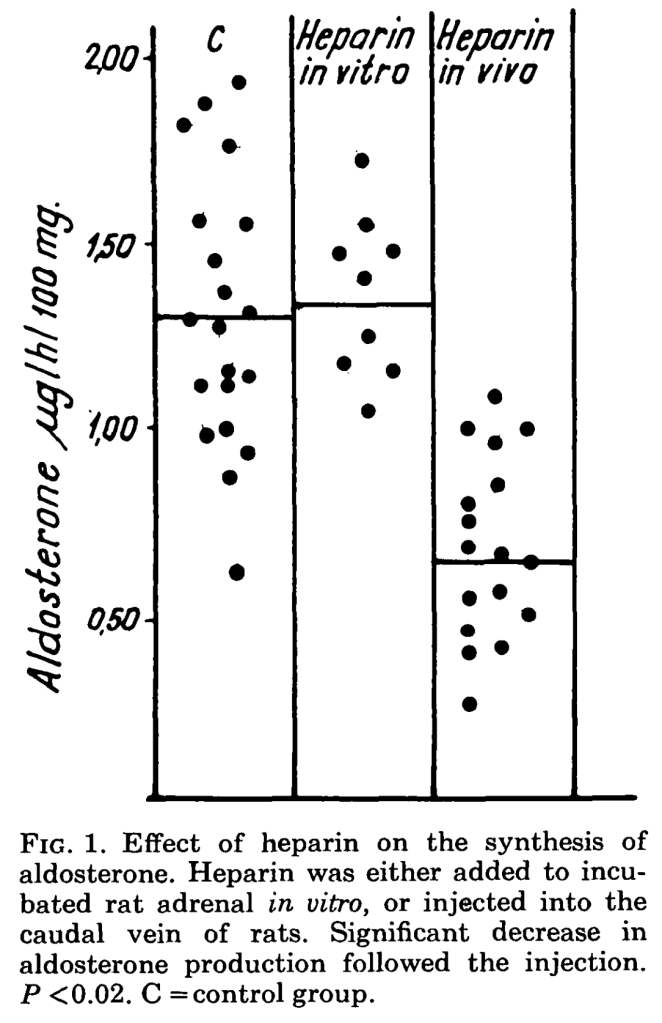
One clue to the mechanism is that heparin reliably reduces adrenal aldosterone production. This was first definitively observed in rats in the 1960s, though clinical studies had suggested this effect even in the 1950s. An experiment in the journal Endocrinology in 1964 found that incubating heparin with rat adrenal gland cells led to a decrease in aldosterone production. A similar effect when was seen when administering heparin to live rats. In both experiments, the decrease in aldosterone was selective. This means that the rat’s adrenal glands only decreased the production of aldosterone after heparin exposure but continued to produce cortisol at normal levels.
Clinical studies in humans have shown the same effect. A study from 1983 in the Journal of Clinical Endocrinology and Metabolism found an approximately 50% reduction in aldosterone during treatment with therapeutic or anticoagulant doses of heparin. A subsequent, smaller study from 1986 in The American Journal of Nephrology found a similar reduction in aldosterone production during treatment with the low doses of heparin that we use for deep vein thrombosis prevention in hospitalized patients.
Why would these effects on aldosterone result in hyperkalemia? Aldosterone is a hormone secreted by the adrenal cortex which activates the eNAC channel and the basolateral Na-K ATPase pump in the distal nephron. Both of these actions lead to the excretion of potassium in the urine. A decrease in aldosterone production from the adrenal glands, therefore, increases serum potassium levels.
Heparin or heparin analogs even seem to suppress aldosterone production in someone with hyperaldosteronism. A case series from 1966 in the Canadian Medical Association Journal reported the results of giving N-formyl chitosan polysulfuric acid (an analog of heparin that has less anticoagulant effects apparently) to two patients with primary hyperaldosteronism and associated hypertension. They found that within two weeks the heparin analog led to a more than five-fold reduction in aldosterone levels in the blood to the point that they couldn’t detect aldosterone in the blood at all. At the same time they noted the patients’ hypertension improved. And, of course, their potassium levels rose.
A review of the renin-angiotensin-aldosterone system, which mediates and controls aldosterone production, is necessary to understand what may be going on. Aldosterone is made from the zona glomerulosa of the adrenal gland. Its production is stimulated primarily by angiotensin II (in addition to some stimulation by adrenocorticotropin hormone or ACTH). Angiotensin II is therefore required to stimulate the adrenal glands to make aldosterone. Heparin directly blocks the effects of angiotensin II at the level of the zona glomerulosa cells in the adrenal gland. This was demonstrated in rats in a study from the Journal of Steroid Biochemistry in 1986. If angiotensin II can’t act on the zona glomerulosa cells, they won’t be able to produce aldosterone.
There are at least potential explanations for this heparin effect. The first is that heparin directly decreases angiotensin II receptor expression on cells of the adrenal zona glomerulosa. This was seen in a rat study in 1988. The second is more theoretical but certainly plausible. There is evidence from mice that heparin blocks angiotensin II-mediated intracellular signaling within vascular endothelial cells, independent of receptor blockade. Heparin disrupted intracellular signaling kinase pathways; this would inhibit angiotensin II’s impact on target cells.
As a result of these effects, heparin has vasodilatory properties, and hypotension is a known potential side effect, particularly when bolused (e.g., in the operating room for cardiac surgery or in the cardiac catheterization lab). Even more fascinating is that heparin has been used as a diuretic. Heparin’s ability to block aldosterone production leads to leads to increased sodium excretion. This data is from the 1960s, before the introduction of loop diuretics. A case series reported the results of using heparin deliberately as a diuretic in 29 hypervolemic patients. Almost all of the patients in the study achieved diuresis while on heparin, with increased urine output and increased urinary sodium excretion. Most of the patients had improvements in body weight and peripheral edema. They highlighted one patient with anasarca from nephrotic syndrome, who lost about 8 kg of body weight over the course of 5 weeks of treatment with intravenous heparin. The patient’s treatment course was interrupted twice for bleeding complications, specifically hemoptysis and hematuria.

Take Home Points
- When you administer heparin products, you’re also administering a drug that has the ability to cause hyperkalemia (though usually mild), vasodilation (particularly when bolused) and potassium-sparing diuretic effects.
- The main mechanisms we discussed were that heparin can decrease adrenal angiotensin II receptor expression, and possibly inhibit intracellular signaling from the angiotensin II receptor, leading to decreased aldosterone production.
CME/MOC
Click here to obtain AMA PRA Category 1 Credits™ (0.5 hours), Non-Physician Attendance (0.5 hours), or ABIM MOC Part 2 (0.5 hours).
Listen to the episode
https://directory.libsyn.com/episode/index/id/24671181
Credits & Citation
◾️Episode written by Avi Cooper
◾️Show notes written by Avi Cooper and Tony Breu
◾️Audio edited by Clair Morgan of nodderly.com
Cooper AZ, Breu AC, Abrams HR. Hyperkalemia Heparin The Curious Clinicians Podcast. October 12, 2022.
Link to related tweetorial: https://x.com/AvrahamCooperMD/status/1444674306135441409?s=20
