

For this episode of the podcast, we discussed why renal failure leads to platelet dysfunction. More specifically we discussed the so-called “uremic platelet”.
Amazingly, the connection between renal failure and bleeding has been observed for a long time. This association was first recognized in 1764 by Giovanni Battista Morgagni. In 1836, Richard Bright observed that purpura frequently occur in uremic patients. In the subsequent 70 years, there were so many additional observations on the topic that David Reisman was able to write a detailed review of them in 1907.
Recent clinical data supports these ancient observations. One study of heart failure patients showed that chronic kidney disease was associated with increased risk of bleeding, with hazard ratios between 1.6 and 2.9, based on the presence of renal replacement therapy.
Where’s the lesion?
Patients with uremic bleeding typically have normal platelet counts, normal aPTTs, and PT/INRs. But uremic bleeding is correlated with prolongation of the skin bleeding time. This is a classic test used to assess platelet function. That it is abnormal in uremia strongly argues for a qualitative platelet problem.
In order to understand what uremia does to platelet function, we need to review primary hemostasis. There are a number of steps involved in the formation of the initial platelet plug. First, endothelial injury leads to collagen exposure. Next, von Willebrand factor (vWF) binds to the newly exposed collagen. Third, platelet glycoprotein Ib (GPIb) binds to vWF. This is known as platelet adhesion. Finally, GPIIb-IIIa crosslinks platelets and also binds vWF. This is platelet aggregation.
Uremia can affect multiple aspects of primary hemostasis. It reduces adhesion (i.e., attachment of platelets to non-platelet surfaces), aggregation (i.e., platelet-platelet clumping), and secretion of platelet factors. Uremic platelets also appear to have a problem storing key factors like ADP, serotonin, TxA2. On the podcast, we focused on problems with adhesion and aggregation.
One study took blood from patients with ESRD just before dialysis and found reductions in platelet adhesion and aggregation. When normal platelets were placed in uremic plasma, their function suffered too. The conclusion: some dialyzable “toxin” in the plasma must be poisoning the platelets.
Urea, creatinine, or something else?
Uremia is named for urea and it certainly accumulates in renal failure. Although there is conflicting data, the consensus is that urea is NOT the toxin that causes uremic platelet dysfunction. One study published in 1970 added urea to normal platelets. They continued to function just fine. There is also a disease called familial azotemia, characterized by high plasma urea but normal renal function. These patients do not experience uremic symptoms and have normal platelet function.
If it isn’t urea, what is the toxin? Guanidinosuccinic acid, or GSA. The same study from 1970 which exonerated urea found that adding GSA to normal platelets made them severely dysfunctional.
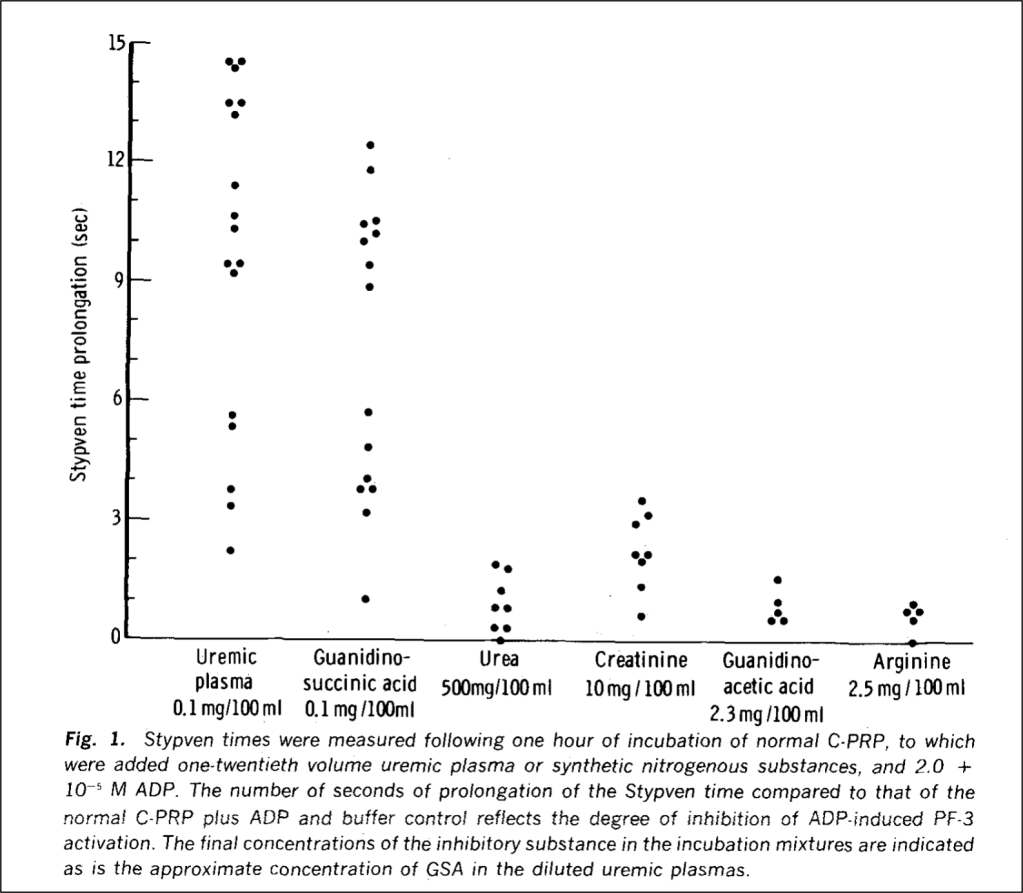
The unheralded GSA
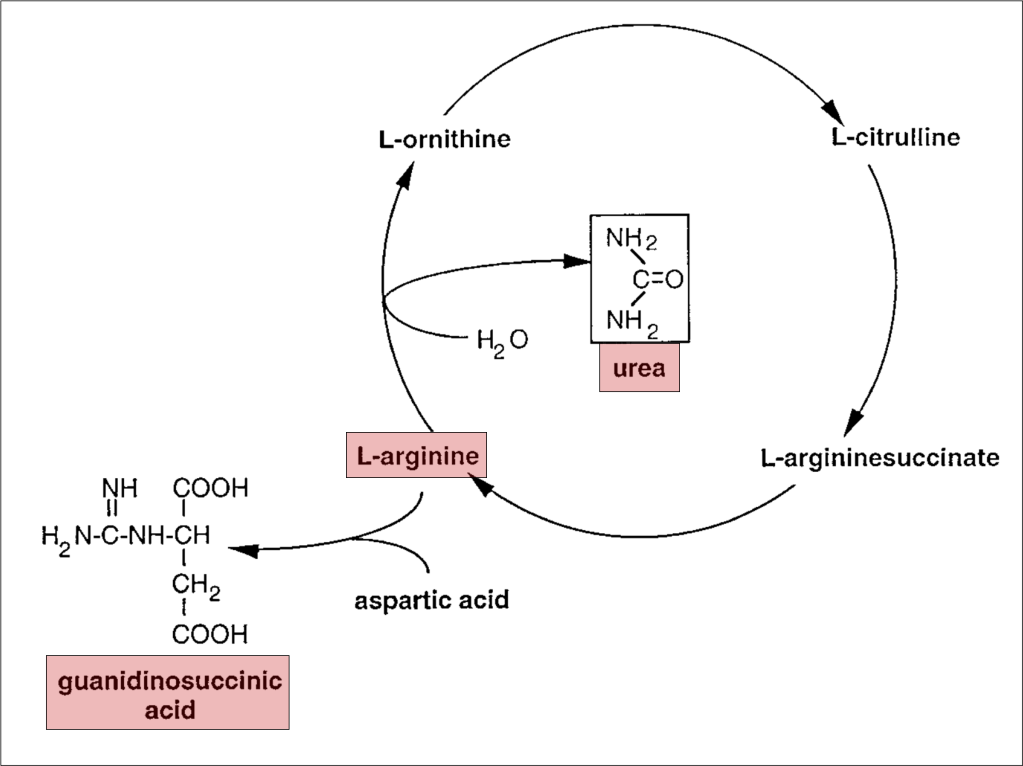
To understand the role of GSA in platelet dysfunction, we’ll have to review what happens to urea normally and in renal failure. Recall that proteins are metabolized to amino acids, which are further metabolized to ammonia. Ammonia is toxic, the metabolism of which is the goal of the urea cycle: it converts highly toxic ammonia to far less toxic urea. The final step in this process is the conversion of L-arginine to urea, which is then excreted in urine.
In renal failure, urea accumulates in the blood. As a result, the enzymes that convert ammonia to urea are down-regulated. As an alternative pathway to deal with toxic ammonia, L-arginine is instead converted to GSA and its serum levels rise.
NO!!!!!
So how does GSA lead to platelet dysfunction?
First, note GSA and L-arginine have similar structures. As a result, one might expect that GSA could be a precursor to molecules for which L-arginine is a precursor. One such molecule is nitric oxide (NO).
We all know that NO is a vasodilator. In fact, the initial endothelial injury that prompts primary hemostasis impairs the ability of endothelial cells to produce nitric oxide. This is one reason why we see vasoconstriction in the setting of endothelial injury. But NO also directly reduces platelet adhesion to the endothelium and platelet-platelet aggregation.
So, to put it all together:
- In uremia, urea accumulates
- In the setting of elevated urea levels, some ammonia is instead converted to GSA
- GSA then accumulates and, given its similarly to L-arginine, is then converted to NO
- NO causes vasodilation AND uremic platelet dysfunction.
There are a few other interesting connections between GSA and uremic platelets:
- Estrogens are an effective treatment for uremic bleeding The proposed mechanism? Estrogen-mediated reduction in nitric oxide!
- GSA may have a role in neurotoxicity as well.
Take Home Points
- Guanidinosuccinic acid (GSA) accumulates in renal failure
- Increased GSA leads to increased nitric oxide levels
- In addition to being a vasodilator, NO has multiple anti-platelet effects
- In short: GSA (via NO) is the uremic toxic that leads to uremic platelets
To read more, here is a Tweetorial on the topic, posted by Tony:
CME/MOC
Click here to obtain AMA PRA Category 1 Credits™ (1.00 hours), Non-Physician Attendance (1.00 hours), or ABIM MOC Part 2 (1.00 hours).
Listen to the episode
Credits & Citation
◾️Episode written by Tony Breu
◾️Audio edited by Clair Morgan of nodderly.com
◾️Show notes by Tony Breu
Breu AC, Cooper AZ, Abrams HR. Why does uremia increase bleeding risk? The Curious Clinicians Podcast. November 11, 2020. https://curiousclinicians.com/2020/10/28/episode-11-why-does-magnesium-treat-torsades-de-pointes/
Image credit: Thomas SG. The Structure of Resting and Activated Platelets. Platelets. 2019:47-77.
