
In this episode, we discussed why calcium is used to “stabilize the cardiac membrane” in hyperkalemia. We covered the evidence for this practice and the proposed mechanisms involved.
As with many topics, a bit of history can be helpful.
Way back in 1883, Sidney Ringer was developing intravenous fluids, including the one that bears his name. He observed that increasing potassium content led to progressively weaker ventricular contractions. To understand what hyperkalemia was doing to the heart, one must first understand the generation of an action potential.
The generation of an action potential is dependent on a number of things, including:
(1) A resting membrane potential, which is approximately −90mV for myocytes.
(2) The threshold potential, which is about -70mV.
(3) The activation state of membrane sodium channels that open during Phase 0 depolarization. The more sodium channels that are open, the more rapid the depolarization.
Alteration of any of these factors might make it easier – or harder – to generate an action potential. Returning to the resting membrane potential, this results from the outward leak of potassium. This leak of positively charged ions leaves the inner membrane relatively negative, typically around -90mV.
If there is more potassium in the extracellular space, the flow of potassium from inside to outside will decrease. And this will make the resting potential LESS negative. So, instead of being -90mV, maybe it’ll change to -80mV. And remember that the threshold potential is -70mV. So, hyperkalemia will initially lead to a membrane that is closer to the threshold.
The result is increased myocyte excitability. You might even say the membrane is “unstable”.
Enter calcium
There are at least two ways that calcium might “stabilize” this unstable – aka excitable – membrane. The way to think about these two mechanisms is to imagine the difference between resting and threshold potentials. This is normally about 20mV. Hyperkalemia might narrow this to around 10mV. If the resting and threshold potentials are closer to each other it is easier to generate an action potential. This is what we mean by unstable or excitable. So what we’re looking for is a therapy like calcium to restore the normal difference of 20mV. It could theoretically do this in one of two ways.
First, it might undo the change in resting potential. If hyperkalemia moves the resting potential from -90mV to -80mV, one possibility is that calcium returns the resting potential back to (or closer to) -90mV. Second, calcium might increase the threshold potential, pushing it away from the resting potential and close to the normal difference of 20mV.
In 1955 Silvio Weidmann showed that MORE depolarization was required in calcium-rich solutions. The resting potential was unchanged, but the threshold potential had increased. Weidmann concluded that a change in threshold potential accounted for the ‘stabilizing’ effect of calcium. It was the second mechanism that mentioned above that was key.
There’s more to it
Calcium does more than just reinstate the difference between resting and threshold potential. And this second effect may be more important. Although hyperkalemia initially makes the resting potential less negative leading to INCREASED membrane excitability, a persistent change in resting potential inactivates sodium channels required for Phase 0. Recall that activation of sodium channels is required for depolarization. So inactivation of these channels leads to DECREASED membrane excitability.
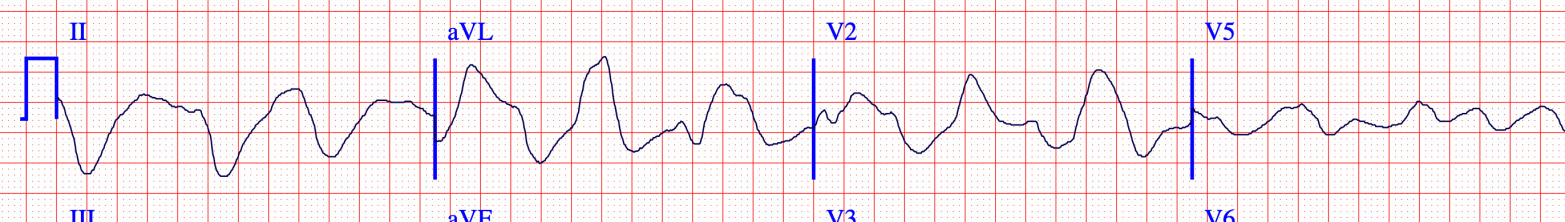
The decrease in membrane excitability is shown by a decreased rate of rise of phase 0 of the action potential. This is the Vmax. We see evidence of this in the widened QRS on the ECG.
So initially the membrane is more excitable or unstable. But persistent hyperkalemia leads to a decrease in excitability. And it may be that the decrease in excitability that is more problematic in hyperkalemia. It fits with the key symptoms of weakness, heart block, and asystole.
Why does hyperkalemia lead to a decreased rate of rise of phase 0? Amazingly, the resting potential during depolarization actually determines the number of active sodium channels. At -90 mV – again this is the normal resting potential – the maximum number of channels are open. As the resting potential moves closer to 0, fewer channels are open leading to slower depolarization. As discussed earlier, hyperkalemia is exactly a scenario where the resting potential is increased.
And calcium protects against this second mechanism too by increasing the activity of the sodium channels required for Phase 0 depolarization. More specifically, calcium salts bind to calcium-dependent calmodulin and protein kinase II and activate these sodium voltage gate channels.
Is there evidence?
The evidence for calcium isn’t as definitive as one might expect. One of the earliest studies is from 1939. That year, Winkler, Hoff, and Smith reported on experiments where they examined the effects of various mixtures of calcium and potassium salts on the heart. When potassium chloride (KCl) was given without calcium, the serum potassium at which death occurs was lower than the values if calcium was added alongside the KCl. This was offered as evidence of the protective effects of calcium.
Over the subsequent decades, other small studies showed similar effects. Not every study was positive, but there were enough cases that calcium became the standard of care. None were randomized trials.
What about sodium?
Since at least 1918, hypertonic saline has been shown effective at combatting the cardiotoxic effects of hyperkalemia. Just as with calcium, hypertonic saline can increase the velocity of Phase 0 depolarization via activation of fast sodium channels. The simplest way to think about this is that dumping a lot of sodium into the extracellular space will increase the velocity of sodium flow across the cell membrane. Hypertonic saline can have similar effects to those seen with calcium. For example, you can see improvement in the ECG abnormalities caused by hyperkalemia. And in reviewing the literature, I am surprised how easily calcium became the standard of care. Before the 1960s, saline and calcium were vying for primacy and reviews mentioned both therapies. Then, two reviews in the 60s – one in The Lancet and one in the NEJM – endorsed calcium. The definitive paper wasn’t published that decade. Instead, this seems to reflect the authors’ opinions and possibly what had become the standard of care.
A brief history of the use calcium and sodium for the treatment of hyperkalemia can be found here.
Take Home Points
- Initially, hyperkalemia increases the resting potential making myocytes MORE excitable
- The increased resting potential inactivates sodium making myocytes LESS excitable
- Calcium salts address both of these issues
- Hypertonic saline may also be beneficial
CME/MOC
Click here to obtain AMA PRA Category 1 Credits™ (1.00 hours), Non-Physician Attendance (1.00 hours), or ABIM MOC Part 2 (1.00 hours).
Listen to the episode
https://directory.libsyn.com/episode/index/id/19243127
Credits & Citation
◾️Episode and show notes written by Tony Breu
◾️Audio edited by Clair Morgan of nodderly.com
Breu AC, Abrams HR ,Cooper AZ. Why do we give calcium in hyperkalemia? The Curious Clinicians Podcast. May 26, 2021
Image credit: https://ecg.bidmc.harvard.edu/maven/mavenmain.asp
Related tweetorial: https://x.com/tony_breu/status/1332717836213628938?s=20

Hi. I have always thought it the other way round…That hyperkalemia generally means inability to return K into the cells. So the resting membrane potential becomes more negative due to far less positive charge inside the cell than outside, hence less excitability of the cells. Consequently we have Brady, weakness, asystole, heart block etc as complications. In this case, calcium with its strong Nernst potential should shuttle quick into the cells once administered via the T-tubules and cause calcium-induced calcium release from the endoplasmic reticulum that leads to a resetting of the resting membrane potential returning excitability that then opens up sodium channels leading up to quicker depolarization but also better muscle contractility.
Regarding hypertonic saline, could it be a result of sudden myocardial stretch with increased myocardial activity per Sterling…
LikeLike
Loved this episode
Would love to be added on your mailing list
LikeLike
I’m a bit late to the conversation but I wanted to comment on an earlier reply. I think the premise that hyper K is an inability to return K to the cells that results in a more negative resting potential is not correct. The negative resting potential is a result of more positive charge existing outside the cell than within, making for a negative membrane potential. It’s true that most of the K in our body is intercellular and most Na is extracellular.
However, what occurrs within the cardiac myocyte is a complex dance of ions and interplay of ion channels. At rest the myocyte is not permeable to Na, so very little Na enters the cell. (Focusing more on ventricular myocytes for simplicity) However, K can slowly leak out of the cell through K leak channels. Therefore, the cells are basically always permeable to K. While at rest, this means a bunch of Na outside and a bunch of K inside, with some of that K leaking out. This bit of K that leaks out means more positive charge outside. This is further enhanced by the NaK pumps that continuously work to maintain this gradient. Hence, the resting potential is always negative (More + charge outside than inside).
If we add a bunch of K to the extracellular environment, it’s easy to see that you now have more positive charge than normal and this negative membrane potential increases or becomes less negative, not more negative. The difference between positive charge inside the cell and outside is essentially converging. Therefore, this pushes the resting potential upwards and away from the -90mv.
Initially, this can lead to hyper excitable myocytes, that are easier to depolarize but sustained hyper K causes certain types of Na channels to close, resulting in slower depolarization and decreased Vmax despite the cell being easier to depolarize. This also results in the prolongation of the action potential and refractory period.
LikeLike